
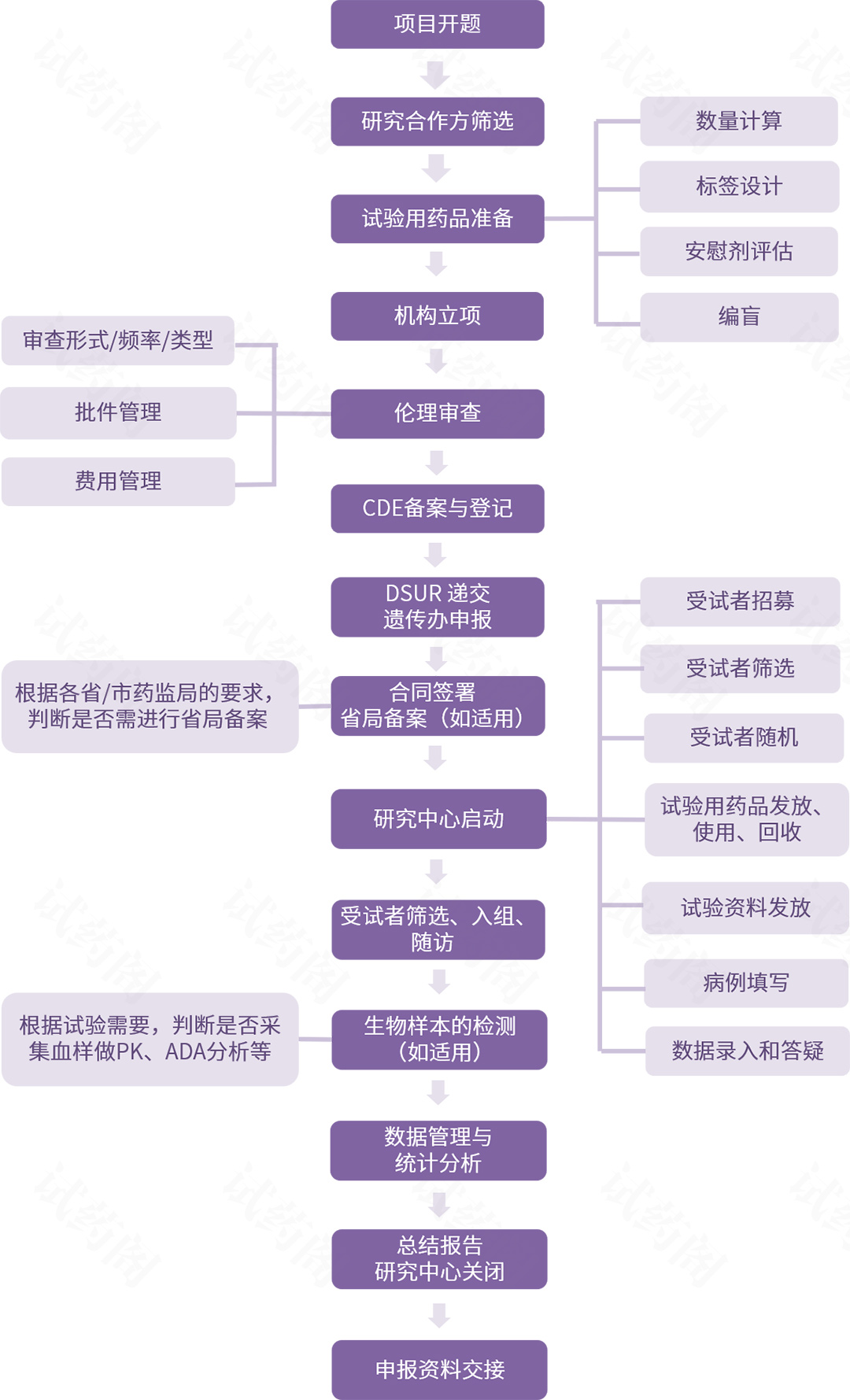
临床试验申办方流程分为三个阶段:启动阶段、实施阶段、总结阶段
1. 临床试验启动阶段
1.1 获得国家药监局的药物临床试验批件(国家药品监督管理局药品审评中心CDE网站注册审批,有效期3年)
1.2 制作研究者手册
研究者手册的内容包含:序言、目录、化学和物理性质、临床前研究、药理学研究、药代动力学研究、毒理学研究、已有临床资料、药品使用信息等。研究者手册是临床试验开始前的资料汇编。
1.3 筛选主要研究者
在NMPA临床试验机构备案系统中,筛选医院(一般来说不是整个医院而是某个科室),然后选择合适的主任级医生;联系主任医生(看是否有意向,在给予任何资料前,签保密协议,然后发放可行性问卷,收集可行性问卷,确认完整无误,回复感谢信,感谢其参与问卷调查)。
注:先拜访研究者还是医院相关部门视具体情况和医院要求而定,没有统一标准。
如果可行,可安排访视,具体考察该中心是否有能力执行临床试验;
(电话预约研究者,给予方案和研究者手册,Agenda确认要讨论的具体问题,如:研究人员的分配、方案的可行性、能否入够病人及如何保证其依从性、药品管理、档案管理、研究者职责、主要研究者资质的确认、相关研究人员的资质、硬件设施考察、伦理委员会的访视(承不承认中心伦理、伦理开会频率、送审清单、伦理委员会人员清单)、书写访视报告、再次拜访,与其讨论方案、试验时间、试验费用等问题。)
1.4 试验文件准备
会同CRO、申办者、主要研究者,共同制定临床试验方案、病例报告表、知情同意书样本、原始文件;CRO制定临床试验中其他用表;
1.5 其他研究者筛选
1.5.1 从NMPA临床试验备案系统中选择其他可能的研究者,可根据首研者的推荐以及公司曾经合作的情况进行综合判断;
1.5.2 准备资料:方案、研究者手册、知情同意书样本、病例报告表;
1.5.3 与其谈论方案,要求提供其医院所能提供的病例数、时间进度和经费预算;
1.5.4 选定合适研究者,征得其医院管理部门的同意。
1.6 召开多中心临床方案认论会(研究者会议),讨论试验方案、CRF等;
注:是否需要召开研究者会议要看具体情况
1.7 取得伦理委员会批件
注:IRB(Independent Review Board)/TEC(Independent Ethics Committee)——IRB为美国的伦理委员会,IEC/C为欧盟的伦理委员会。
按照GCP的要求,所有临床试验必须得到伦理委员会的批准。在实际进行临床试验时,首先必须取得牵头单位伦理委员会的批准,其他参加单位是否要过伦理根据各家单位的具体要求而定。
伦理委员会将对有关批件、药检报告、研究者手册、知情同意书样本、试验方案(protocal)、病例报告表进行审批;
1.8 试验药品准备
督促申办方进行试验用药品的送检;生物统计师设计随机分组方案;根据随机分组方案,设计药品标签;设计应急信件;
药品包装:为每一个受试者准备好一盒药物,药盒上贴好标签,并装入相应的应急信件;
1.9 各种物品印刷和准备(CRF、知情同意书、患者日记、标签、药盒、礼品)
注:1.7及1.8可在研究者会议后开始准备,同时申请伦理委员会批件。
1.10 各方签订协议
1.11 召开临床试验启动会(启动访视),事后书写访视报告
会议内容包括:①试验人员培训,以达到统一标准的目的,②试验相关文件、表格、药品分发到各研究者。
1.12 在临床试验登记平台进行登记
1.12.1 药物临床试验登记与信息公示的范围和内容
凡获国家药监局临床试验批件并在我国进行临床试验(含生物等效性试验、PK试验/药物代谢动力学试验、I、Ⅱ、Ⅲ、IV期试验等)的,均应登陆信息平台(网址:http://www.chinadrugtrials.org.cn/index.html),按要求进行临床试验登记与信息公示。
登记内容包括:①临床试验批件的复印件、②己确定的临床试验方案、③临床试验负责机构及主要研究者姓名参加研究单位及研究者名单、④伦理委员会审核同意书、⑤知情同意书样本、⑥CRF样本
1.12.2 药物临床试验登记与信息公示的实施要求
自本公告发布之日起,对新获得药物临床试验批件的,申请人须在获批件后1个月内完成试验预登记,以获取试验唯一登记号;在第1例受试者入组前完成后续信息登记,并首次提交公示。获批件1年内未完成首次提交公示的,中请人须提交说明;3年内未完成首次提交公示的,批件自行废止。
对已获得药物临床试验批件且批件有效的,申请人须在本公告发布之日起3个月内完成信息登记。
药物临床试验启动后,申请人与研究者应根据相关规范性文件要求与《药物临床试验登记填写指南》,通过信息平台及时完成相关试验信息更新与登记公示。
2. 临床试验实施阶段
2.1 访视前充分准备
2.1.1 制定试验的总体访视时间表
2.1.2 令每一次访视前,回顾试验的进展情况、前次未解决的问题;
2.1.3 与研究者联系,确定访视日期,并了解试验用品是否充足;
2.1.4 制定访视工作的计划、日程表,准备访视所需的文件资料和物品;
2.2 监查项目
2.2.1 与研究者会面说明本次访视的主要任务,了解试验进展情况(受试者入选情况、病例报告表填写情况),以前访视所发现问题的解决情况。
2.2.2 核对并更新研究者管理文件夹,研究人员及职责有无变化(更新研究者列表、新研究者履历、并对其培训),检查并补充试验用品;
2.2.3 研究设施有无变化(是否校正、正常值范围、设施品牌、耗材供应状况)
2.2.4 检查知情同意书(ICF:informed consent form):①签字日期与入选日期;②签名情况(见证人、监护人、医生);③版本号码;④修改日期;⑤新情况发生,是否修改知情同意书(送伦理委员会);⑥是否交予受试者;⑦了解知情同意过程。
2.2.6 收集病例报告表;
2.2.7试验药品管理的核查(存放情况、发放回收情况记录、清点药品并与相应记录核对、检查盲码信封、使用是否违反方案要求):①检查药品的保存状况和记录情况;②检查药品数量,与记录的数量核对;③检查应急信封;④检查药物使用情况的记录(患者日记),是否违反方案要求;⑤是否按随机号码发放。
2.2.8 AE不良事件的处理
①检查SAE(严重不良事件)的报告(报告程序是否符合GCP及标准操作规程(SOP)要求报告及时间,是否通知申办者、NMPA、伦理委员会、其他研究者)和跟踪;
②SAE页填写情况(是否记录了不良事件的种类、描述、开始时间与持续时间、相关症状、轻重程度、发生频度、所做检查和治疗,记录规范、处理是否及时);
③SAE处理(是否得到了应有的医疗保护或适当的经济补偿、是否停药);
④确认是否与试验药物相关;
⑤是否需要开启应急信封;
⑥跟踪不良事件的最终结果;
⑦监查所有不良事件的临床资料,再次查看知情同意书;
⑧注意个人隐私,受试者在试验中的编号,不暴露其姓名、住址和身份证号码。
2.2.9 研究者文件夹的更新
2.2.10 书写监查报告
2.3 记录所发现的问题;
2.4 与研究者一起讨论和解决此次访视发现的问题,交流其他研究单位的进展和经验;
2.5 将取回的药品、物品、已签署的知情同意书、病例报告表等按规定存放;
2.6 填写访视报告;
2.7 更新各项记录表格;
2.8 对发现问题的追踪及解决;
2.9 安排后续访视计划;
2.10 临床试验过程中,如试验方案、知情同意书、病例报告表发生改动,需报伦理委员会审批;
2.11 临床试验中发生SAE(严重不良事件),必须在24小时内报告药品监督部门,并尽快报告伦理委员会和申办者;
2.12 病例报告表收集同时,生物统计师建立数据库,设定逻辑校验程序,并将收集到的病例报告表输入。输入过程中发现病例报告表有问题,则立即提交监查员,由监查员在下次访视中加以解决;
2.13 当数据库中已有一定病例记录时,生物统计师开始编撰统计分析程序,并利用己有数据进行调试。
3. 临床试验总结阶段
结束访视:访视前的准备:电话预约时间并确认
3.1 检查并解决常规访视中遗留问题;
3.2 收集所有病例报告表并与原始文件核对检查;
3.3 通知伦理委员会
3.4 试验用药的回收和销毁(结束访视)
①详细记录试验用药品的回收、存放;
②详细记录临床药品的销毁方法及经过
3.5 回收所有试验用品
3.6 更新所有记录表格
3.7 书写监查报告,档案归档
3.6 数据入库
在进行阶段,已经进行了一遍数据输入,收集所有病例报告表后,再输入一遍。
将两遍输入的数据进行自动校对,输出两者差别表。根据两者差别表,对照原始病例报告表进行改正
3.7 生物统计师编写数据逻辑校验程序,以程序分析数据库中数据的合理性
3.8 对于逻辑校验程序发现的问题,对照原始病例报告表。如果是输入错误,则加以改正;如果是原始病例报告表填写有误,则再返回医院,进行检查更正
3.9 所有数据通过数据逻辑校验程序的审查后,锁定数据库
3.10 统计分析
生物统计师(甲)编写统计分析程序。
对每个医院进行分析;对所有医院总和进行分析;对符合方案集进行分析;对意向集进行分析。
生物统计师(乙)编写验算程序,对生物统计师(甲)的分析结果进行验算。
生物统计师提交统计分析报告。
3.11 召开临床试验总结会(二次揭盲)
3.12 合同尾款结算
3.13 申报资料盖章
3.14 资料准备
CRF等物资(QA、QC、统计)合同(注意小包项目、考核)药品(我方或厂家)备案。
3.15 会同研究者、申办者、CRO,根据统计分析结果,撰写临床试验总结报告
①临床监查员独立或协同研究者起草临床总结报告和分中心小结;
②临床总结报告和分中心小结最终由研究者审核并确定,并交由各中心签字、盖章。
3.16 临床资料存档
临床试验中所有文件均需按GCP 要求存档,并指定与人负责。应严格遵循“No record ,
No action“之原则,对临床中涉及的每项工作均进行文件归档管理,并按照GCP要求存放。
注:GCP与ICH-GCP对文件的归档要求稍有不同,主要是对原件的归档要求不同。这时需要询问每个中心的归档清单,必要时准备两份原件,盖两个章,确保中心和申办者文件夹都有原件。
3.17 向药监局提交临床试验总结和相关文件,注册报批。
3.11 召开临床试验总结会(二次揭盲)
3.12 合同尾款结算
3.13 申报资料盖章
3.14 资料准备
CRF等物资(QA、QC、统计)合同(注意小包项目、考核)药品(我方或厂家)备案。
3.15 会同研究者、申办者、CRO,根据统计分析结果,撰写临床试验总结报告
①临床监查员独立或协同研究者起草临床总结报告和分中心小结;
②临床总结报告和分中心小结最终由研究者审核并确定,并交由各中心签字、盖章。
3.16 临床资料存档
临床试验中所有文件均需按GCP 要求存档,并指定与人负责。应严格遵循“No record.
No action“之原则,对临床中涉及的每项工作均进行文件归档管理,并按照GCP要求存放。
注:GCP与ICH-GCP对文件的归档要求稍有不同,主要是对原件的归档要求不同。这时需要询问每个基地的归档清单,必要时准备两份原件,盖两个章,确保基地和申办者文件夹都有原件。
3.17 向CFDA提交临床试验总结和相关文件,注册报批。
 关注微信公众号
关注微信公众号
