
临床试验中的数据分析不可或缺,在临床试验设计、开展、中间分析和试验完成时分析的各个阶段中,试验数据的处理与分析均需利用统计分析方法展开
1. 分析对象的数据集
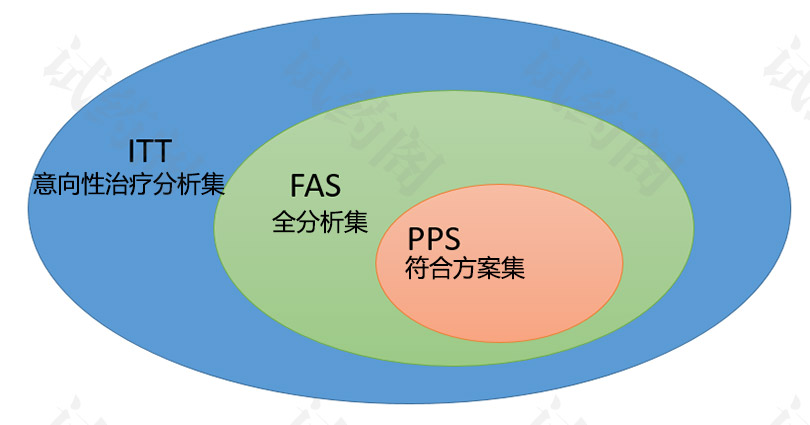
1.1 全样本分析集FAS(Full analysis set)
意向性治疗原则ITT(intention-to-treat)是指主要分析应当包括所有进入随机化的受试者,无论他是否完成治疗
遵循这一原则需要对所有随机受试者完成随访得到试验结果,但由于种种原因,实际上是难以完成的
因此,全样本分析是尽可能接近于包括所有随机受试者,在分析中保留最初的随机化对于防止偏差和提供安全的统计检验基础很重要。
在许多场合,它提供的对治疗效果的估算很可能反映了以后的实际观察结果。
从分析中剔除已随机受试者的情况不多:包括不符合重要入选标准,一次也没有用药,随机化后没有任何数据。
从分析中剔除不符合入选条件受试者必须不致引起偏差:入选标准的测定是在随机化之后;违反合格标准的检测是完全客观的;所有受试者都受到同样的合格性调查;各组实行同样的入选标准,凡违反者均被排除。
1.2 符合方案集PPS(Per Protocol Set)
“Per Protocol”对象组,有时称之为”有效病例”、”有效样本”或”可评价受试者样本;定义为全部分析样本中较好遵循设计书的一个受试者亚组:·完成预先说明的确定治疗方案暴露。·得到主要变量的测定数据。·没有违反包括入选标准在内的重要试验设计。从”有效受试者”组中剔除受试者的精确理由应当在揭盲前就充分限定并有文件记载。为得到”有效受试者”而排除对象的原因和其他一些违反研究设计的问题,包括对象分配错误、试验中使用了试验方案规定不能用的药物、依从性差、出组和数据缺失等,应当在不同治疗组之间对其类型、发生频率和发生时间进行评价。
1.3 不同的分析(受试者)组的作用
在验证性试验中,通常进行全样本和”有效受试者”两种分析。这样可以对两者之间的任何差别进行明白的讨论和解释。有时候可能需要计划进一步探究结论对于选择分析受试者组的敏感程度。两种分析得到基本一致的结论时,治疗结果的可信度增加。但是要记住,需要?quot;有效受试者”中排除相当数量受试者会对试验的总有效性留下疑点。在优越性(Superiority trial,证明新药比标准对照药物优越)试验、等效性试验或不差于(non-inferioritytrial,确证新产品与对照药物相当)试验中,这两种分析有不同的作用。在优越性试验中,全样本分析用于主要的分析可以避免”有效受试者”分析对疗效的过于乐观的估算;全样本分析所包括的不依从受试者一般会缩小所估算的治疗作用。但是,在等效性或不差于试验中使用全样本分析通常是不谨慎的,对其意义应当非常仔细考虑。
2. 缺失值和线外值(包括异常值)
缺失值代表临床试验中一个潜在的偏差来源。因此,在实施临床试验时应当尽最大努力符合试验方案对于数据收集和数据管理的要求。对于缺失值并没有通用的处理办法,但只要处理方法合理,特别是如果处理缺失值方法在试验方案中预先写明,则不会影响试验的有效性。当缺失值数目较大时,要考虑分析结果对于处理缺失值方法的敏感程度。线外值(包括异常值)的统计学定义在某种程度上带有随意性。除了统计学判断之外加上医学判断以鉴别一个线外值(包括异常值)是最可信的方法。同样,处理线外值(包括异常值)的程序应当在方案中列出,且不可事先就有利于某一个治疗组。
3. 数据的类型、显著性检验和可信限
在临床试验中,对每个受试者可收集3种数据:所接受的治疗、对治疗的反应(Re-sponse)和进入试验时影响预后因子的基线值。接受同样治疗的受试者构成统计分疗组。对治疗的反应基本上有3类。 ①定性反应。根据预定的评价标准将受试者分为若干类别,如高血压治疗的”有效”。”无效”;淋巴细胞瘤化疗的”完全缓解”、”部分缓解”、”无变化”。 ②定量反应。当存在一种可靠测定方法时,受试者的治疗结果最好采用实际数值,如舒张压。但最好同时记录其基线值,以便评价治疗前后的变化量值。 ③到某事件发生的时间。如使用避孕药受试者从开始治疗到意外妊娠的时间。
3.1 数据的描述性统计
在开始分析之前,有必要先看一下各组受试者的每个变量观察值的分布频度,以对变量有一个感性了解;从最大值和最小值也可以发现可能的错误和超范围的值;决定某些变量是否需要作某种转换;或按某种特定分布作统计分析。①定性数据需要记录各治疗组的受试者总数和在每个反应类别的受试者数,然后转化为比率或百分率或直方图、圆图等表示。采用c2检验、Fisher精确检验比较所观察到的组间率的差异的程度。②定量数据计算每个治疗组的平均反应(均数、几何均数)和变化程度(标准差)。以均值、标准差、直方图、累积频数分布图表示。在受试者数较小时,可以用图表显示每个受试者的确切反应。组间比较采用t检验、F检验等。当样本值频数图呈偏态分布时,用均值描述定量反应不合适,可采用中位数、四分位数来描述数据的定量水平。组间比较可采用非参数方法。
3.2 显著性检验
显著性检验的真正含义是应用概率理论计算如果两个治疗实际上同样有效时得到所观测到的治疗差异的概率。其目的是评价一个治疗真正优于另一个治疗的证据有多强。这种证据的强度用概率,即P值来定量。因此P值越小,治疗差异由于偶然发生的可能性越小。在实践中,人们常用P<0.05.P<0.01、P<0.001表示显著性检验的结果,这些水平的选择是完全随意的,并没有数学或临床的理由。在解释显著性检验时要注意以下几点:一个小的P值如P<0.05并不是一种治疗优越的绝对证明,每20个真正阴性试验会出现一个假阳性结果;P>0.05也并不证明两治疗同样有效,差异可能实际上存在,只是现有数据不足以证明它存在。统计显著性并不等同于临床重要性,一个10万人的试验中,1%的反应率差异在5%水平是显著的,但在一个20人的试验中40%的差异在统计上也是不显著的。因此,临床的意义必须用差异的大小,即可信限来评价。双侧检验和单侧检验:假设治疗差异可以发生在任一方向时,为双侧检验。双侧检验的零假设为μa=μb;备择假设为μa1μb。如果在试验之前就确定治疗A不可能差于治疗B,为单侧检验。其零假设为μa=μb;备择假设为μa≥μb。此时显著性检验评价A好于B或A相当于B的证据。若结果是A比B差,便归于机遇,因为A不可能差于B。结果是单侧检验的P值为双侧检验的一半。也就是说,单侧检验比双侧检验容易拒绝零假设。采用单侧检验应该有足够的依据。如果试验设计中决定用单侧检验,在结果表示时要注意一般统计软件计算的都是双侧检验的P值。
3.3 可信限的估算
显著性检验只告诉我们一个治疗比另一个好的证据的强度,并没有告诉我们好多少。因此,显著性检验并不是分析的终结,还应运用统计估算方法,如可信限估算治疗改善的量。计算可信限时,应注意被分析变量的统计分布;标准误和可信限的计算方法应该写明。记住必须提供治疗效应大小的统计估算、显著性水平和可信区间。100(1-α)%可信区间,正态分布估算值可表示为{估算值+Nα/2×SE},估算值+(N1-α/2×SE)};差值如呈t分布时,可表示为{x1-x2-(t1-α/2×SEdiff),x1-x2+(t1-α/2×SEdiff)}等。
3.4 对象的基线水平的组间比较
对治疗组的疗效评价只有当各组受试者的基线特征具有可比性时才是有效的。通常,随机化可以提供充分的可比性。但是,随机化并不能绝对保证可比性。有时候组间的基线水平可能会有差异。这种差异对治疗比较的影响应当采用其他程序消除。
3.5 调节显著性和可信限水平
许多情况都可能产生多重性:例如多个终点/主要变量(如血压记录卧位或坐位的收缩压和舒张压;心肌梗死预防试验中的各种原因死亡率和心肌梗死发病率),治疗的多重比较(几个治疗组间比较或试验药物的几个剂量组),及不同时间点的多次测定和中期分析等。存在多重性时,检验主要假设的次数增加,产生I类错误的机会就会变大。分析数据时可能有必要对五类错误进行控制和调节。首先,最好能避免或减少多重性的产生,如从多个主要变量中鉴别出关键的主要变量(如血压记录取卧位舒张压为主要变量;心肌梗死预防试验取死亡率为主要变量);对反复测定则采用一个综合测量指标如”曲线下面积”。多重比较的常用统计方法有Bonferroni方法、Holm法和Hochberg方法。Bonferroni方法是一个保守的方法,对于成对比较,它调节P值以控制总的I类误差率。Hochberg方法比另两种方法更有效,它只需控制最大的P值小于显著性水平。多个终点的α调节用Bonferroni方法和Hochberg方法。
3.6 亚组、相互作用和协变量
除了治疗以外,主要变量常与其他影响系统相关。主要变量可能与协变量如年龄和性别有关;或在受试者亚组之间可能存在差异,如多中心试验中在不同中心接受治疗。在某些情况下,调节协变量影响或亚组效应是所计划的分析的一个必要部分。要特别注意中心的影响和主要变量的基线测量值的作用。不要在主分析中对随机化以后测定的协变量进行调节,因为这些测定可能受治疗的影响。此外,治疗效果本身也可能随亚组或协变量改变。疗效可能随年龄而下降,或在具有某一特殊预后因子的受试者中增大。这类相互作用在某些情况下是可以预见的,或具有特殊的意义(如老年病学),因此,一个亚组分析或包括相互作用项的统计模型是所计划的验证性分析的一个部分。对于定量反应变量,多元回归是最常用的统计调节方法,有时也称协方差分析。对于定性反应,可以应用多元Lgistic模型。
3.7 评价安全性和耐受性
3.7.1 评价范围
一个药物的有用性总是在风险和效益之间的平衡。在所有临床试验中,安全性和耐受性评价是重要内容之一。在临床研究早期阶段,这类评价带有探索性,仅注意毒性的表达方式;在较后阶段,则是在大样本对象中更全面地确定药物的安全性和耐受性特征。后期的对照临床试验是以一种无偏倚方式揭示任何新的不良反应的重要手段,尽管此类试验的把握度有限。
3.7.2 变量选择和数据收集
在临床试验中,选择评价药物安全性和耐受性的方法和测定取决于一系列因素:药物不良反应的知识,药物非临床研究和早期临床试验以及重要的药效学/药代动力学特征资料,给药方案,被研究对象和研究持续时间。安全性和耐受性的主要数据通常包括临床化学和血液学的实验室测试(如WBC、SGPT),生命指征和体检(如血压、ECG),临床不良事件(疾病、体征和综合症)。发生严重不良事件和因不良事件中断治疗对于注册是特别重要的数据。临床试验中使用共同的不良事件编码词典特别重要。这种词典的结构提供了在3个不同的水平总结不良事件数据的可能性:系统-器官分类,标准术语(preferred term)和包括术语(included term)。通常,不良事件按标准术语分类总结,相同系统-器官分类的标准术语在数据的描述性报告中可以放在一起。现在常用的有世界卫生组织的《疾病和有关健康问题的国际统计分类》ICD-10,和美国的COSTART。
3.7.3 评价的受试者和数据报告
安全性和耐受性评价中,所总结的受试者通常至少曾接受过一个剂量研究药物。要尽可能全面地从这些受试者中收集安全性和耐受性变量,包括不良事件的种类、严重程度、开始时间和持续时间,以及处理方法和结果。评价时要注意所有安全性和耐受性变量。所有不良事件,不管它们是否与治疗相关,都应当报告。实验室测定值的单位和正常范围应有明确定义。使用的毒性分级标度(toxicity grading scale)应当预先说明。通常一个特定不良事件的发生率表示为经历该事件受试者数相对于处于危险的受试者数的率。但是,根据需要,被暴露的受试者数或暴露程度(用人-年表示)可以作为分母。不管其目的是为了估算危险度还是在治疗组间进行比较,应该在方案中明确定义,这在计划长期治疗并预期会有相当比例的治疗中止或死亡时特别重要。在这种情况下,应当考虑采用生存分析(Survival Analysis),计算不良事件累积率以避免低估危 当存在明显的症状或综合征基线噪声时,估算不良事件危险度的一个办法是采用”治疗引发”(treatment emergent)概念,只记录与治疗前基线相比时原先没有的不良事件或症状变重的不良事件。减少基线噪声的其他办法还有:不计轻度的不良事件,一个事件在重复随访中观察到才计算。不论采用何种方法,都须在方案中说明理由。
3.7.4 安全性的统计评价
在大多数临床试验中,安全性和耐受性结论的陈述多采用描述性统计方法,辅以有助于解释的可信区间计算。用图可表示治疗组内不良事件的类型。计算P值有时也是有用的:可以评价一个事件的差异,或是在大量安全性和耐受性变量中突出值得进一步注意的差别。计算P值对于总结实验室数据特别有用。实验室数据可进行两种分析:评价均值的定量分析和计算高于或低于某一个阈值的数目定性分析。
 关注微信公众号
关注微信公众号
