
临床试验是指,在人体(患者或者健康志愿者)进行药物的系统性研究,流程一般包括:立项前、立项、伦理、遗传办、合同、中心启动、临床观察、临床试验结束这8个步骤,目的是确定药物的安全性与有效性
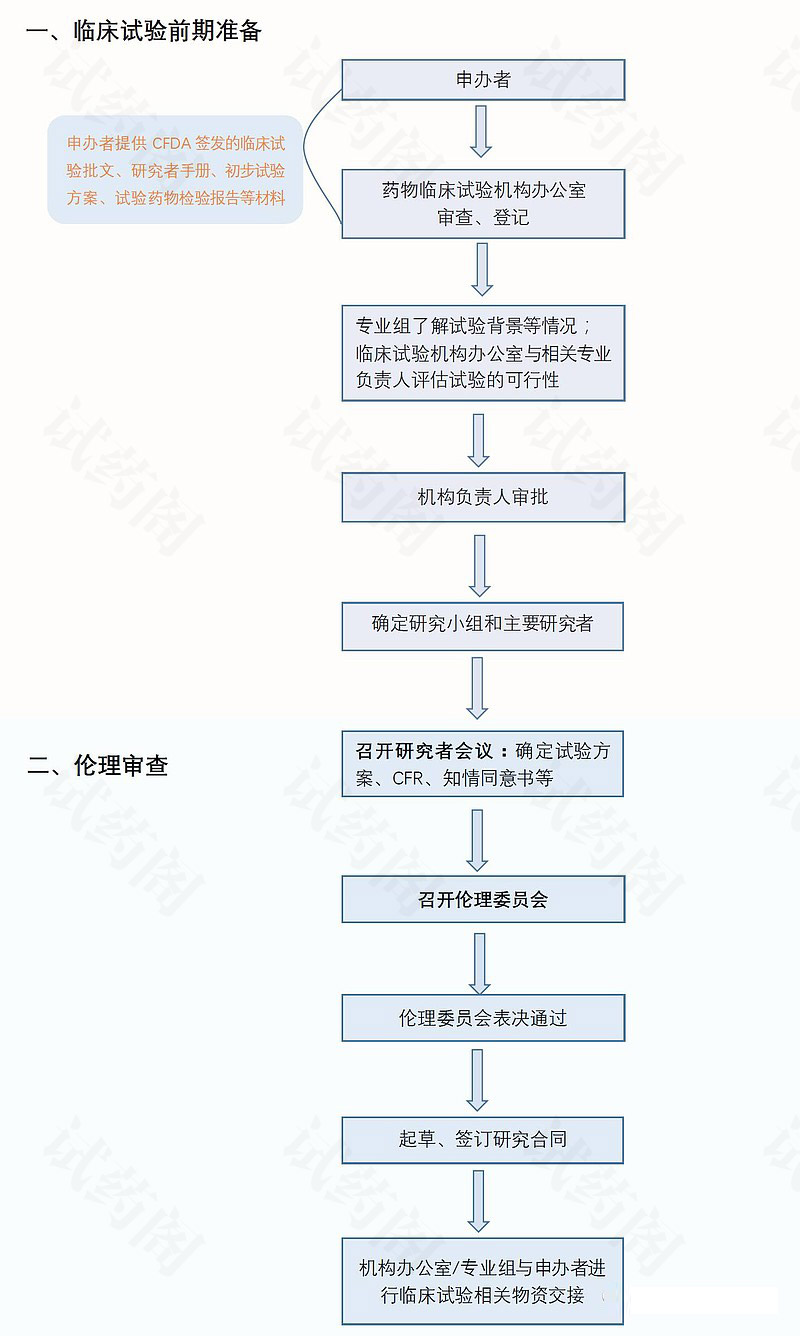
1.立项之前
- 申办方发起临床试验。→获取国家药监局批件
- 筛选研究中心(调研)。→项目承接科室及PI→执行者:CRO/SMO
-
研究者会议前准备。→执行者:CRO/SMO
- a.发送研究者会议邀请函:PI/Sub-I 机构;
- b.收集研究者行程时间、身份证号码、联系方式;
- c.协助研究者顺利往返会议,收集发票。
- 召开研究者会议。确定方案、监查、稽查、IB、SOP、职责分工、CRF、ICF等。
2.立项
- 向临床试验机构递交立项申请。 →执行者:CRO
- 形式-邮件/CTMS系统
- 内容-立项申请表、国家局批件、方案、IB、保密协议、PI资质、职责分工表、申办方资质
- 机构进行形式审查。→执行者:CRO/SMO
- 符合要求后方可递交纸质机构立项资料夹备案:需PI签字的原件,盖章要求:骑缝章、公章,递交信最好一式2份。
- 机构召开立项会。
- 给出意见→口头或电话通知,或者CTMS系统:主委或者项目负责人CTMS系统签字。
- 立项成功,进入伦理申请阶段。
3.伦理递交
•伦理委员会:由医学专业人员、法律专家、社区及非医务人员组成的独立组织,其职责为核查临床试验方案及附件是否合乎道德,并为之提供公众保证,确保受试者的安全、健康和权益受到保护。该委员会的组成和一切活动不应受临床试验组织和实施者的干扰或影响。
•伦理委员会至少5人,应有不同年龄层次和不同性别的委员
- 向伦理递交伦理初始审查申请。 →执行者:CRO
- 形式–邮件/CTMS系统
- 内容–伦理初始审查申请表、国家局批件、方案、IB、申办方资质、组长单位批件、ICF、日记卡、保险等
- 伦理进行形式审查。符合要求后方可递交纸质机构立项资料夹备案,需PI签字的原件,盖章要求:骑缝章、公章,递交信最好一式2份。
- 伦理进行上会通知。–提前了解上会的频率、时间及费用及时跟进
- 上会并获取伦理意见。–同意、不同意、作必要修正后同意
- 领取批件。–注意核对药物类别
- 按照中心流程开取发票,邮寄至CRA处。–伦理收据、开票信息及打款证明
4.遗传办申请
- 递交遗传办承诺书至机构。
- 同意组长单位批件,签署后领取承诺书。
- 若不同意组长单位批件,需分中心单独申请,具体参照文档。
- 获取遗传办批件。
注:同时可收集实验室质检质控证明、正常值范围、相关检查费用、研究者简历及相关资质;
5.合同签署
- 根据机构要求准备合同模板。
- CRA/CRC确定合同份数、签署所需时间、骑缝章及公章
- (机构审核合同时间:一般是获取伦理意见后)
- 申办方、机构及PI 三方共同审阅合同并确认合同内容。
- (涉及研究费用、试验相关检查费用及试验用设备的收集,设备主要有药品/样本冰箱、温度计、离心机、打印机、电脑、文件柜)
- 及时跟进合同审核、签署情况。
- 必要时协助CRA递交合同
- 合同签署后及时领取。
- 邮寄CRA/申办方
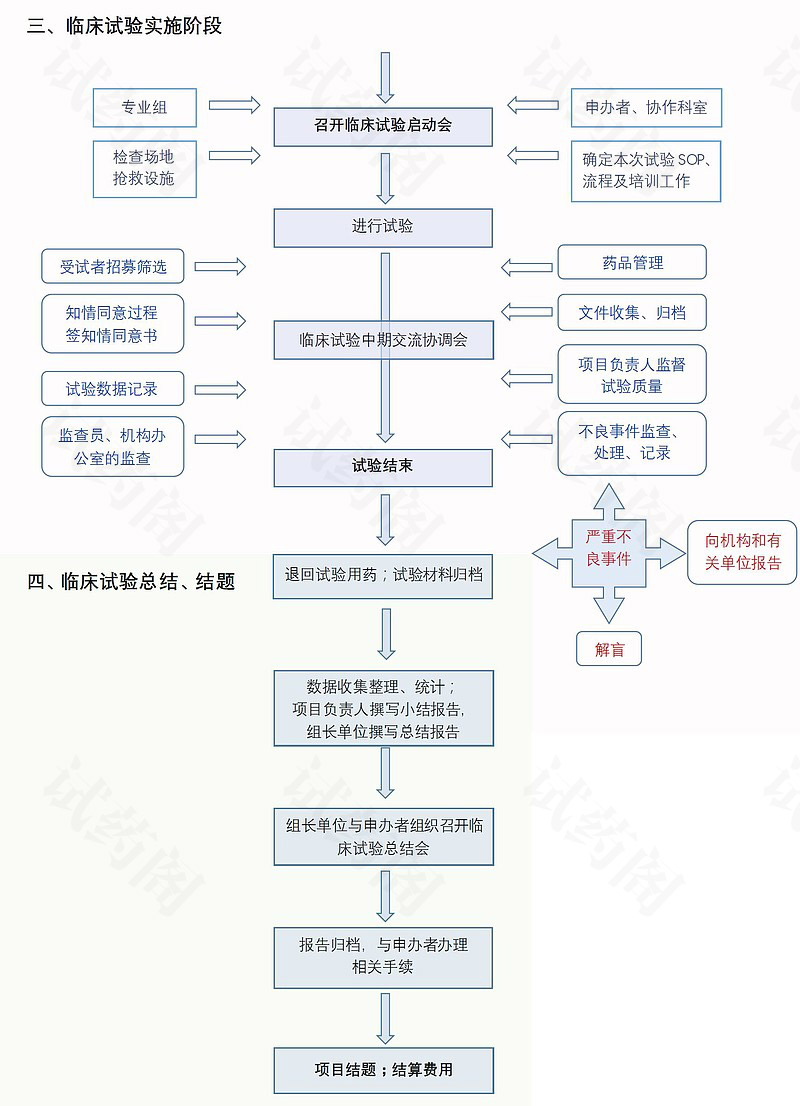
6.中心启动
- 中心启动前。 CRC/CRA
- a.确认人员分工,文件、物资、设备、药品已运送至中心。文件:方案、ICF、伦理批件、已签署的研究协议、已签署的方案签字页、研究者简历等。提前调试好药物储藏冰箱,符合药物保存要求。
- b.确认会议时间、地点及餐食及主要人员、文具、PPT、简单入排卡。
- 召开启动会。
- 会场准备需要完成的表格:授权表、培训记录、会议签到表。
- 跟进解决启动会研究者所提问题。
- 试验正式开始。
7.临床观察
- 严格按照入排标准筛选受试者。逐条筛选排除(一票否决法)
- 筛选成功入组 注意原始病历及病史的收集,每条记录需要原始文件的支持
- 持续进行AE/SAE及合并用药的收集、EDC录入。
- CRA监查、申办方稽查、国家药监局视察、项目组内一级质控、科室内二级质控及三级机构质控。
- 递交外院备案SAE、方案更新件等资料。
8.临床试验结束
- 试验药物退回
- 合同尾款结算
- 专业组质控
- 机构质控(锁数据库之后,项目负责人根据统计结果撰写小结报告,组长单位撰写总结报告)
- 专业组提交小结报告给CRA和CRC确认内容,机构秘书确认内容完整性
- 专业组提交小结报告给伦理审核
- 桶过伦理后专业组召开结题会议
- 会议后机构办资料管理员整理、归档文件资料,制作文件管理目录
- 完成药物临床试验结题确认表,小结报告盖章2份,一份交申办者,一份归档
- 机构办公室提交财务报告,机构主任审批,财务科发放劳务费或统一划帐
- 总结报告的归档,再次整理和保存
 关注微信公众号
关注微信公众号
